I've recently embarked on a study to see what effect clustering mass spectra has on the design of MRM experiments. Specifically one clusters spectra from the shotgun proteomics discovery phase of the experiment to see how it affects the selection of peptide species and the transitions that a mass spec looks for during the second targeted sequencing phase of the experiment.
While the work is ongoing, a very interesting (at least to me) result has come out of plotting the unfiltered SEQUEST results from the non-clustered and clustered version of the mass spec data. The say a picture is worth a thousand words, so:
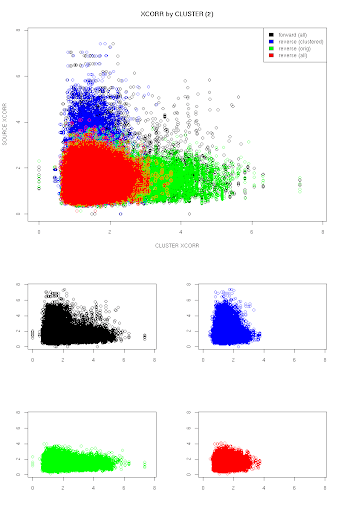
The chart is showing the XCORR values of the clustered spectra plotted against the XCORR values for their respective members, independent of what peptide was identified for each. The color coding represent scores for peptides that are part of the decoy database, where:
Particularly, it is going to be very interesting to see whether clustering actually helps or hinders ion selection when designing MRM experiments. If it does help ion selection, I have already come up with the catchy name, Heuristically Beneficial Artifacts TM. If not, well, we will at least have looked at the real-world effects of a methodology when applied to a new type of experimental goal.
More on this as it develops.
While the work is ongoing, a very interesting (at least to me) result has come out of plotting the unfiltered SEQUEST results from the non-clustered and clustered version of the mass spec data. The say a picture is worth a thousand words, so:

The chart is showing the XCORR values of the clustered spectra plotted against the XCORR values for their respective members, independent of what peptide was identified for each. The color coding represent scores for peptides that are part of the decoy database, where:
- blue = a decoy hit was scored in the clustered spectra, but not the original
- green = a decoy hit was scored in the original spectra, but not the clustered spectra
- red = both spectra scored a decoy peptide
Particularly, it is going to be very interesting to see whether clustering actually helps or hinders ion selection when designing MRM experiments. If it does help ion selection, I have already come up with the catchy name, Heuristically Beneficial Artifacts TM. If not, well, we will at least have looked at the real-world effects of a methodology when applied to a new type of experimental goal.
More on this as it develops.
